-
La maladie en chiffres
Le processus de détérioration se produit lors d’un dépôt excessif de collagène et autres protéines du tissu conjonctif, provoquant une cicatrisation progressive du tissu pulmonaire par l'inflammation et la fibrose.Ce remodelage tissulaire désorganisé compromet le rôle vital des poumons dans sa fonction respiratoire, avec des conséquences dévastatrices en termes de capacité fonctionnelle, de qualité de vie et de mortalité accrue.1
La prévalence des PI a été estimée à 81/100 000 chez les hommes et 67/100 000 chez les femmes.2Parmi elles, la plus fréquente est la fibrose pulmonaire idiopathique (FPI). En Europe, la prévalence de la FPI varie entre 1,25 et 23,4 cas pour 100 000 habitants.3
En outre, les maladies interstitielles pulmonaires touchent 15 % des patients atteints de pathologies du tissu conjonctif (CTD). Il s’agit d’un groupe de maladies auto-immunes dont la plus courante, la polyarthrite rhumatoïde (PR), touche entre 0,5 et 2 % de la population aux États-Unis.4
![]()
Visionnez notre vidéo pédagogique
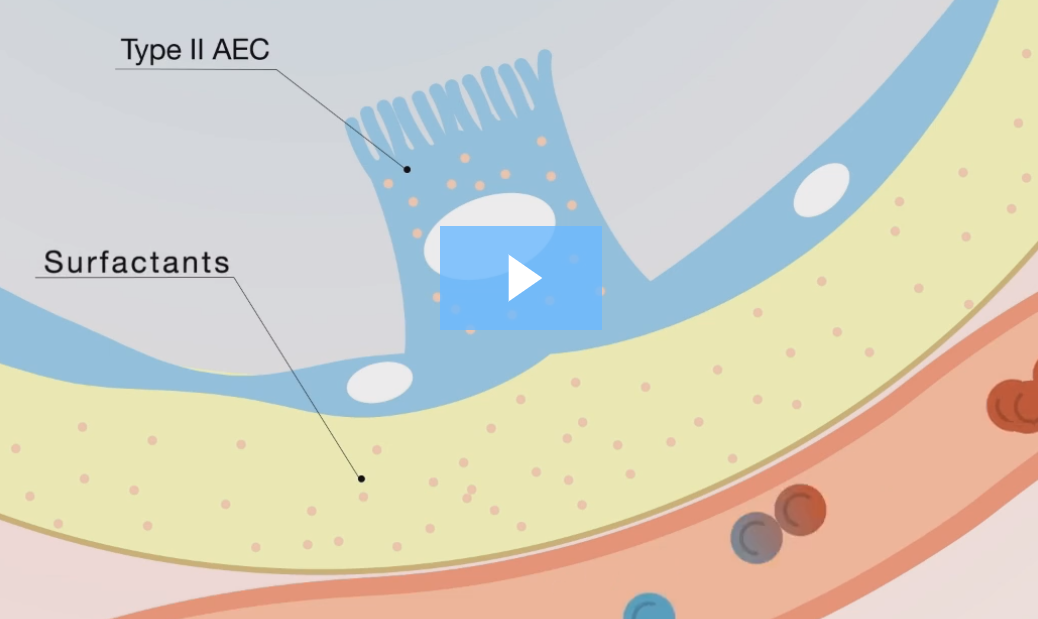

Pneumopathies Interstitielles (PI)
Des millions de personnes souffrent de maladies pulmonaires affectant différentes parties du système respiratoire, comme les voies aériennes, les alvéoles, l'interstitium, les vaisseaux sanguins et la plèvre.
Un groupe important et hétérogène de maladies dénommées pneumopathies interstitielles (PI) ou pneumopathies parenchymateuses diffuses affectent l'interstitium. Il s’agit d’une fine couche cellulaire inter-alvéolaires contenant des vaisseaux sanguins et des cellules soutenant l’architecture des alvéoles, permettant ainsi un échange gazeux efficace.
