-
La prevalencia de las EPI se estima en 81/100.000 en hombres y 67/100.000 en mujeres.2
Entre las EPI, la clasificación más común es la Fibrosis Pulmonar Idiopática (FPI). En Europa, la prevalencia de FPI se estima entre 1,25 y 23,4 casos por cada 100.000 sujetos.3
Además, la EPI aparece en un 15% de los pacientes con enfermedades tejido-conectivas (ETC), que es un grupo de enfermedades autoinmunes, de las cuales la más frecuente, la artritis reumatoide (AR), afecta entre un 0.5% y un 2% de la población general en USA.4
![]()
Mire nuestro video educacional
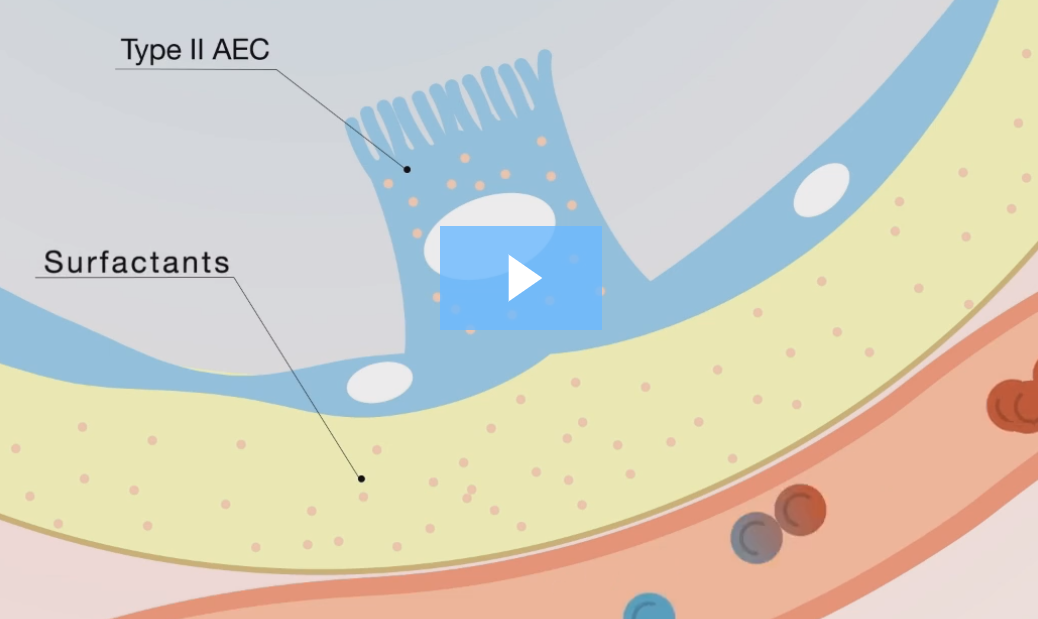

Enfermedades Pulmonares Intersticiales (EPI)
Millones de personas padecen de enfermedades pulmonares que pueden afectar cualquier parte de su sistema respiratorio, incluyendo las vías respiratorias, los alveolos, el intersticio, los vasos sanguíneos y la pleura.
Un grupo grande de enfermedades conocidas como Enfermedades Pulmonares Intersticiales (EPI) afectan el intersticio, una fina capa de células entre los alveolos, que contiene vasos sanguíneos y células que ayudan a los alveolos, permitiendo un intercambio de gases eficiente.
